
Was sind die Ursachen für Familiärer Hypercholesterinämie?
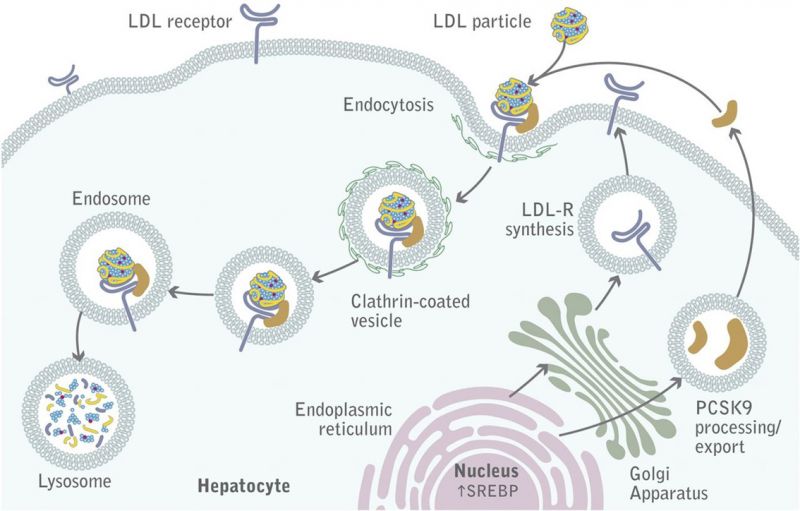
Die Familiäre Hypercholesterinämie ist genetisch bedingt und wird in 85-90 % der Fälle durch eine Mutation im Gen des LDL-Rezeptors (LDL-R) ausgelöst. Eine Verminderung der Zahl oder Störung der Funktion der LDL-R hemmen die Aufnahme und den Abbau des LDL-Cholesterins und steigern in der Folge die Cholesterinsynthese in der Leber. Das führt zu erheblichen Erhöhungen des LDL-C im Plasma und zu Ablagerungen in den Gefäßen oder im Gewebe. In Mutationsdatenbanken sind aktuell mehr als 1.700 Mutationen des LDL-R gelistet.
Neben Mutationen des LDL-R können genetische Defekte des Apolipoprotein B-100 und der Protease PCSK9 (Proprotein convertase subtilisin/kexin type 9) Erhöhungen des LDL-Cholesterins verursachen.
Diese drei bekannten Ursachen der FH sind phänotypisch, d. h. sie sind klinisch, nicht eindeutig unterscheidbar. Aufgrund dieser Erkenntnis wurde der Begriff autosomal dominante Hypercholesterinämie (ADH) oder auch heterozygote Familiäre Hypercholesterinämie geprägt.
Davon abzugrenzen ist die seltene autosomal rezessive Hypercholesterinämie oder auch homozygote Familiäre Hypercholesterinämie, für die zwei Defektallele des LDL-R-Adapterproteins 1 (LDLRAP1) verantwortlich sind.
In wenigen Fällen einer diagnostizierten Familiären Hypercholesterinämie kann in keinem der drei Proteine eine Veränderung gefunden werden. In diesen Fällen ist der Auslöser vermutlich eine Veränderung in einem anderen Protein, dessen Rolle für den Cholesterinstoffwechsel nach bisherigem Wissenstand noch nicht bekannt ist.
© The American Society for Biochemistry and Molecular Biology – Die Abbildung zum „LDL-Stoffwechsel“ wurde im Original in the Journal of Lipid Research, December 2012 vol. 53 no. 12 pp. 2515-2524 veröffentlicht.